Bloom Sendromu: Belirtileri, Nedenleri ve Tedavisi
Bloom sendromu (BS), başlıca üç yönü ile karakterize nadir görülen otozomal resesif kalıtım hastalığıdır: büyüme geriliği, güneşe aşırı duyarlılık ve yüzdeki telenjiektazi (kapiler damarların genişlemesi). Bu hastalar, kolayca kanser geliştirmelerine neden olan genomik bir dengesizliğe sahiptir.
Dermatolog David Bloom tarafından 1954'te, cücelik ve telanjiektatik eritem (kan kılcal damarlarının genişlemesi nedeniyle kızarık cilt) sunan birkaç hastanın gözlenmesiyle keşfedilmiştir (Elbendary, 2015).
Bu sendrom aynı zamanda konjenital telangiektatik eritem veya Bloom-Torre-Machacek sendromu olarak da adlandırılabilir.
Bloom sendromunun nedenleri
Bloom sendromu otozomal resesif bir hastalıktır, yani, hem anneden hem de babadan BLM geninin her iki alelinde bir mutasyonun olması gerekir (Ellis ve ark. 1995). Ebeveynler mutlaka bu hastalığa sahip olmak zorunda değillerdir, ancak belirtileri olmadan mutasyona uğramış genin taşıyıcıları olabilirler.
Bloom sendromunda BLM geninde 60'dan fazla mutasyon bulundu, en sık 2281 pozisyonunda 6 nükleotidin silinmesi ve 7 başka birinin ikamesi (Elbendary, 2015).
Genetics Home Reference (2016) 'ya göre, BLM geni, helikas ailesinin bir parçası olan RecQ proteini oluşturulmasına yönelik talimatların gönderilmesinden sorumludur.
Helikaslar, DNA'ya katılmak ve çoğaltılması (veya DNA kopyasını), hücre bölünmesi ve onarımı için hazırlıkları yapmak gibi işlemleri geliştirmek amacıyla, genellikle spiral şeklinde birbirine bağlanmış iki şeridini geçici olarak ayırmaktır. DNA hasarı.
Kısacası, RecQ helikazları DNA'nın yapısını korumak için önemlidir ve bu nedenle "genom bakıcıları" olarak bilinir.
Örneğin, bir hücre iki yeni hücre oluşturmak için bölünecekse, kromozomlardaki DNA'nın kopyalanması gerekir, böylece her yeni hücrenin her bir kromozomun iki kopyası olur: babalardan biri ve annenin diğeri.
Her bir kromozomdan kopyalanan DNA, kardeş kromatitler adı verilen iki özdeş yapıya sahiptir ve hücrelerin bölünmesi gerçekleşmeden önce başlangıçta bağlanırlar.
Bu aşamada, aralarında bazı DNA parçaları değiş tokuş ederler; kardeş kromatid değişimi olarak bilinen şey. BLM proteini zarar gördüğünden ve bu, kardeş kromatitler arasındaki doğru değişimleri kontrol eden ve DNA'nın kopyalanma sırasında sabit kaldığı için bu işlem Bloom hastalığında değişmiş gibi görünüyor.
Aslında, Bloom sendromunda kromatitler arasındaki normal değişimlerden ortalama 10 daha fazlası vardır (Seki ve ark. 2006).
Öte yandan, bu hastalıkta genetik materyalde de kopmalar vardır, bu da normal hücresel faaliyetlerde BLM proteininin eksikliğinden dolayı onarılamayacak şekilde bozulmaya neden olur.
Aslında, bazı uzmanlar bu sendromu "kromozom kopma sendromu" olarak sınıflandırır, çünkü çok sayıda kopma ve kromozomun yeniden düzenlenmesi ile ilgilidir.
Kromozomların bu dengesizliği, hastalıkların gelişmesi olasılığını arttırır. Örneğin, BLM proteininin eksikliğinden dolayı, ultraviyole ışığına neden olabilecek DNA hasarından kurtulamazlar ve bu nedenle bu hastalar ışığa duyarlıdır.
Ek olarak, etkilenenler, onları enfeksiyona daha duyarlı hale getiren immün yetmezliğe sahiptir.
Öte yandan, kontrolsüz hücre bölünmesi ile herhangi bir organda kanser gelişme olasılığı yüksektir, bunlar genellikle lösemi (beyaz kan hücrelerinin fazlalığı ile karakterize bir tür kan kanseridir) ve lenfoma (sistemin lenf düğümündeki kanser) görünür. bağışıklık).
Ayrıca, DNA hasarını onarmaya yarayan MM1 ve MM2 proteinlerinin kodlanmasından sorumlu olan FANCM geninin etkisinde de başarısızlıklar bulunmuştur.
Bunlar hem bu sendromla hem de Fanconi anemisiyle bağlantılı olanlar. Bu yüzden bu iki hastalığın fenotiplerinde ve hematolojik tümörlere yatkınlıkta ve kemik iliğinde yetersizlikle aynı olduğunu görüyoruz.
Her neyse, Bloom sendromundaki kromozomları etkileyen moleküler mekanizmalar halen araştırılmaktadır.
Prevalansı nedir?
Bloom sendromu nispeten nadirdir, sadece tıbbi literatürde açıklanan yaklaşık 300 vaka bilinmektedir. Bu rahatsızlık birçok etnik grupta ortaya çıksa da, Aşkenazi Yahudilerinde bu sendromlu hastaların% 25'ini oluşturan daha yaygın gibi görünüyor.
Aslında, bu etnik grup içinde sendromu ortaya koyma sıklığı% 1'e ulaşabilir. Japon ailelerinde de, daha az sıklıkla olsa da bulunmuştur.
Cinsiyete gelince, erkeklerin hastalığa yakalanma olasılığı kadınlardan biraz daha fazla, bu oran 1 kadın için 1.3 erkek.
Belirtileri nelerdir?
Bu durum yaşamın ilk aylarında zaten ortaya çıkmıştır ve şu anda hiçbir hasta 50 yıldan fazla yaşamamıştır.
- Malign tümörler : yukarıda açıklandığı gibi genomik kararsızlığın neden olduğu, bu sendromdan etkilenenlerde ölümün ana nedenidir. Ulusal Nadir Bozukluklar Örgütü'ne (2014) göre, Bloom sendromundan etkilenenlerin yaklaşık% 20'si kanser geliştirecek. Bu hastalar, bu bozukluğu olmayan insanlardan 150 ila 300 kat daha fazla kanser geliştirme riskine sahiptir.
- Hastaya göre ciddiyette değişen ve çeşitli enfeksiyonlara yatkın olan immün yetmezlik . Bu, lenfositlerin (beyaz kan hücreleri) çoğalmasındaki eksiklikler, immünoglobülin (bağışıklık sisteminin antikorları) sentezindeki problemler ve (hücrelerin bölünmesini ve büyümesini kontrol eden) mitojenlerin uyarılmasına düşük yanıt nedeniyle ortaya çıkar.
- T ve B lenfositlerindeki kusurlar, bağışıklık sisteminin gelişimini etkileyen sık görülür.
- Bağışıklık sisteminin arızalanması kulak enfeksiyonu (başlıca otitis media), zatürree veya diyare ve kusma gibi diğer belirtilerle sonuçlanabilir.
- Işığa duyarlılık: DNA'nın ultraviyole ışınlarına aşırı duyarlılığı, zarar görmesidir. Güneş çarptığında etkilenen cildine zarar veren bir fototoksisite veya hücre ölümü şekli olarak kabul edilir.
- Doğurganlığın veya kısırlığın azaltılması . Aslında, erkeklerde bekleme üretme yetersizliği vardır. Kadınlarda çok erken bir menopoz vardır.
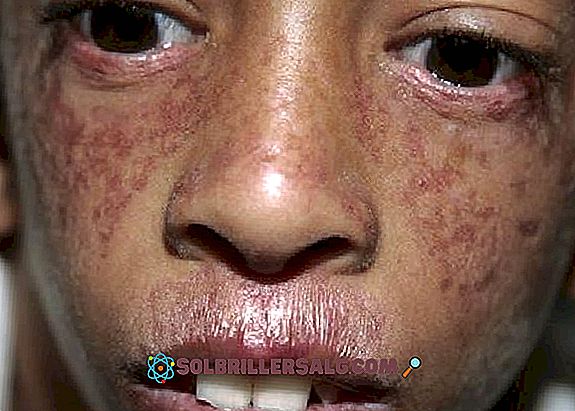
- Kutanöz tezahürler : ışığa duyarlılığa ek olarak, poikiloderma da ortaya çıkar, cildin esas olarak boyunda ortaya çıkan, hipopigmentli bölgelere, diğer hiperpigmentli bölgelere, telenjiektazi ve atrofiye neden olan bir enfeksiyonu. Deride (özellikle yüzünde) güneşe maruz kalma ile ilişkili kırmızı noktalar görülür.
- Gözlenen bir diğer kutanöz problem, küçük kan damarlarının genişlemesinden kaynaklanan yüzdeki kırmızımsı püskürmeler olarak görülen telanjiektazidir . Burun ve yanakları örten bir "kelebek" deseni gibi görünür.
- Vücudun diğer bölgelerinde de anormal kahverengi veya gri lekeler görülebilir ("café au lait" lekeleri).
- Zaten bebeklerde ortaya çıkan gelişimdeki gecikme . Küçükler genellikle normalden daha dar ve daha küçük, kendine özgü bir kafa ve yüze sahiptir.
- Etkilenenlerin yaklaşık% 10'u diyabetin gelişmesine neden olmaktadır (Ulusal Nadir Bozukluklar Örgütü, 2014).
- Çok keskin bir ses .
- Dişlerdeki değişiklikler .
- Gözlerdeki anormallikler, kulaklar (belirgin kulaklar), eller veya ayaklar (hasta normalden daha fazla parmağa sahip olduğunda meydana gelen polidaktili gibi).
- Pilonidal kistler.
- Beslenme sorunları : Özellikle bebeklerde ve küçük çocuklarda fark edilir, yemeğe ilgi duymadığını gösterir. Birçok kez ciddi gastroözofageal reflü eşlik eder.
- Entelektüel kapasiteler değişkendir, bu nedenle bazı hastalarda daha fazla kötüleşir, diğerlerinde ise normaldir.
Nasıl teşhis edilir?
Aşağıdaki testlerden herhangi biri ile teşhis edilebilir:
- Kromozomal sapmaları ve kardeş kromatitlerin değişim seviyesini ölçen sitogenetik testler .
Kanda kültürlenen lenfositlerde dört radyal birlikteliklerin (dört kol kromatitlerin değişimi) varlığını gözlemlemek mümkündür, herhangi bir hücrede, kromatid boşluklarında, kırılmalar veya yeniden düzenlemelerde yüksek miktarda kardeş kromatid değişimi olup olmadığını kontrol etmek; Veya, BLM geninde mutasyon olup olmadığını doğrudan görün.
Bu testler, BLM genindeki mutasyonları taşıyan ve onları yavrularına aktaran sağlıklı bir birey tespit edebilir.
Amerika Birleşik Devletleri Gıda ve İlaç İdaresi (FDA) Şubat 2015'te genetik testin bu hastalığın varlığının erken tespit edilmesinde yararlı olabilecek "23veMe" tarafından ticarileştirildiğini açıkladı.
Bu klinik koşullar varsa, bu sendromun varlığından şüphelenilmelidir:
- Rahim içi dönemden bu yana görülen önemli büyüme gecikmeleri.
- Güneşe maruz kaldıktan sonra yüzün cildinde eritemlerin varlığı .
Şaşırtmayın ...
Bloom sendromunu teşhis etmeden önce onları ekarte etmek için aşağıdaki sendromlar dikkate alınmalıdır:
- Kromozomların kırılması ve yeniden düzenlenmesiyle ilişkili diğer otozomal resesif kromozomal instabilite sendromları, konuyu, özellikle, diğer genleri içeren ve BLM'yi içermeyen Fanconi anemi, ataksi telangiektazi veya kseroderma pigmentoza gibi belirli kanser türlerine karşı hassas hale getirir .
- Genç yaşta gecikmiş gelişim, ışığa duyarlılık ve yaşlanma görünümü ile kendini gösteren kalıtsal bir hastalıktan oluşan Cockayne sendromu. Bu nadir görülen bir cüceliktir.
- Rothmund-Thomson sendromu : Oldukça nadir görülür ve tipik cilt anormallikleri, saç kusurları, genç kataraktlar, kısa boy ve kraniyofasiyal malformasyonlar gibi iskelet değişiklikleri ile kendini gösterir. Bloom'un cilt iltihabı, poikiloderma, cilt dejenerasyonu (atrofi) ve telanjiektazi sendromuna benzer.
tedavi
Bloom sendromu için, yani aşırı mutasyon sayısı için spesifik bir tedavi yoktur. Aksine, müdahaleler semptomları hafifletmeyi, destek sunmayı ve komplikasyonları önlemeyi amaçlamaktadır.
- Kendinizi doğrudan güneş ışığına maruz bırakmamaya çalışın.
- Yeterli güneş kremi kullanın.
- Cildin lekelerini, kızarıklığını ve iltihabını tedavi etmek için bir dermatolog tarafından takip edin.
- Enfeksiyonlar için antibiyotik kullanın.
- Özellikle bu hastalar yetişkinliğe ulaştığında olası kanser vakalarını tespit etmek için düzenli tıbbi kontroller. Olası semptomlara dikkat etmeye çalışmalıyız, çünkü iyileşmeleri için erken cerrahi olarak çıkarılması gereken tümörler vardır. Kanserin erken teşhisi için bazı yöntemler mamografi, Papanicolaou veya vajinal sitoloji veya kolonoskopidir.
- Bu çocukların sindirim reflüsüne müdahale etmeye çalışan gerekli besinleri aldıklarını kontrol edin. Bunun için, uyurken tamamlayıcı beslenme için bağırsak sisteminin üst kısmına bir tüp yerleştirilebilir. Bu, az miktarda yağ birikimini biraz artırabilir, ancak büyümenin kendisi üzerinde bir etkisi olduğu görünmüyor.
- Diyabet varlığını en kısa sürede tedavi etmek için inceleyin.
- Birey kanseri varsa, kemik iliği nakli tasarlanabilir.
- Aile ve diğer destek grupları ve benzer hastalıkları olan dernekler, böylece etkilenen birey, mümkün olan en yüksek yaşam kalitesine sahip bir kişi olarak gelişir.
- Eğer ailede bu hastalık vakası ya da eşin ailesi tarafından bulunmuşsa, genetik danışma tıbbi karar vermeye katkıda bulunmak için bu tür rahatsızlıkların niteliği, kalıtım ve sonuçları hakkında bilgi edinmede faydalı olacaktır. ve kişisel.